
17th Annual Presidential Lecture Series
https://doi.org/10.17077/7a09-e5b1
Genes and Environment:
Science and Society
Professor of Pediatrics,
Pediatric Dentistry, and
Biological Sciences
The University of Iowa
Introduction
First, I would like to thank Professors Coelho and Nosikova for the beautiful introductory. Next, it is imperative to thank President Coleman and the selection committee for this very particular and great honor. While it is a particular honor, it is not, of course, singular, and I hope over the course of this presentation to identify many of the people whose collaboration and collegiality have enabled me to be in the position of delivering this talk to all of you today. As you know, the title is “Genes and Environment, Science and Society,” and what I hope to outline for you is both the scientific basis for the work that we do and our accomplishments so far, as well as the broader implications for society of not just our work but of studies of genes and environment in general.
The mandate for these talks was outlined by President Freedman in 1983:
To present significant aspects of his or her scholarly work to the entire university community and thereby stimulate intellectual communication among the many disciplines that comprise the university.
At the first presidential lecture in 1984, Sherman Paul captured the essence of the mission in two brief questions:
What am I thus privileged to say? What have you come here to hear?
While it is impossible for me to know what you have come to hear, I hope that many of you will have an interest in science and, in particular, in the powerful new approaches of the Human Genome Project to providing an understanding of the role that genes and environment play in human disease. The implications of this genetic research then, for not only our own society here at the University, in Iowa City, in the State of Iowa and in the United States, but more importantly for the world as a whole, are broad, and often unanticipated. I hope to provide you with a bit of a personal view as to what some of these implications might be.
My motive in speaking to you today is best captured by Jacob Bronowski in his 1956 book, Science and Human Values. Dr. Bronowski characterized the work of a scientist as follows: “What a scientist does is compounded of two interests: the interest of his time, and his own interest.” This does, indeed, well capture what I will outline today.
As a final introductory comment, let me state for those of you in the audience, and I hope there are some, at the beginning of your careers, whether in high school, college, graduate school, or even early in your professional careers, I hope that if nothing else, you can realize from what I outline here that for me, as well as for you, it is a great privilege to live in this country and in this time. With hard work, intelligence and some luck, it is possible to not only work on what interests you, the kinds of things that drive you to stay up late into the evening, reading and thinking about problems, about questions, to establish close personal friendships with individuals who share a similar passion, but also to have that work be something of value to science as a whole. While few of us can work on the most important questions of the time and while most certainly my own work is unexceptional in comparison to the important problems facing us as a human society today, it does provide for the opportunity to make incremental improvements in the lives of people everywhere. No matter what your chosen field, if you can both enjoy your work and feel satisfaction that lives of people somewhere have been bettered by the work that you’ve done, whether through the work itself or even through your personal contact with your fellows, you can consider your life work to be a success.
I will outline three scientific opportunities that have resulted from our work. First, I will describe our work on the Human Genome Project, an extensive collaborative effort that involved many individuals here at the University, as well as around the world.
Next, I will talk about how we have applied some of these developments to our own specific interests in diseases that cause birth defects. I will describe our successful identification of a particular gene that causes inherited eye and tooth abnormalities, our ongoing work to better understand how cleft lip and palate, a common birth defect, results from a combination of gene and environmental influences, and finally, how we have been able to extend these studies from outside of Iowa to international populations where the work itself has both greater urgency and opportunity.
Lastly, I will try to synthesize how these particular projects and the broader ones they represent raise issues of morals, ethics and values for all of us.
Scientific Considerations of the Human Genome Project
Our work with the Human Genome Project began while I was a postdoctoral fellow in Seattle, Washington and developed an interest in human genetic mapping. In the early 1980’s, it had become apparent that the identification of variations in DNA sequence, or so-called polymorphisms, would enable scientists to map genes with unprecedented power. This work had been presaged by the identification in 1953 of the molecular structure of the DNA molecule by Watson and Crick described here in the opening paragraph of their article:
We wish to suggest a structure for the salt of deoxyribose nucleic acid (D.N.A.). This structure has novel features that are of considerable biological interest.
Almost 50 years later, as the genome project was beginning its work, Erwin Chargraff—the biochemist whose description of the A=T, G=C role allowed Watson and Crick to build their DNA model—described current efforts as follows:
This world is given to us on loan. We come and we go; and after awhile we leave earth and air and water to others who come after us. My generation, or perhaps the one preceding mine, has been the first to engage, under the leadership of the exact sciences, in a destructive colonial warfare against nature. The future will curse us for it.
These two quotations capture both the enthusiasm, excitement and promise of the genome project as outlined by Watson and Crick and the concerns of many about the potential for terrible effects and outcomes that might also result from this work.
The genome project itself has as its very core the promise of delivering gene therapy or the treatment of genetic disorders with magic genetic bullets that could potentially correct even the most serious birth defects or disorders of aging, such as Alzheimer’s disease, Parkinson’s, cardiovascular disease, diabetes and many others. It also holds out the promise of cloning, whether that be individual organs for replacement parts in humans or an entire individual from a single cell, as occurred with the sheep, Dolly.
The reality of the Human Genome Project is different, and at the present time, affords us improvements in diagnostic capability, particularly the ability to identify a predisposition to a disorder such as breast cancer or Alzheimer’s disease before it begins to manifest itself clinically. In addition, the genome project is delivering tremendous advances in technology, particularly in biology, as it is often only through gene identification and discovery that the involvement of pharmacologists, biochemists, anatomists, embryologists, physiologists and others can begin to determine the best mechanism by which treatment and prevention can be undertaken.
Genome—the genetic component of an individual or species
I’ve yet to define genome, and it is necessary to do so, for the word itself can be used in at least two important ways. First, the human species, as is true of all species, has its own genome: that is, the invariant nature of the DNA sequence which contains the information necessary for making and constructing from a fertilized egg an individual of that particular species. At the same time, each of us, unless an identical twin, differs from one another, and many of those differences result not just from changes in the environment in which one grows up, but rather from the very underlying nature of the genetic material itself and how it can vary from person to person. These variations include common differences, such as those found in skin, hair or eye color, as well as the rare differences which may underlie a predisposition to a particular disorder.
Identical twins, by virtue of the fact that they appear to be exact copies of one another, tell us that our physical appearance and structure is almost, if not entirely, genetically determined—a bold statement, but one that certainly can be supported by looking at two individuals who share all of their genetic material in common. Of course, it is equally true that even identical twins do not share a large number of personality traits, interests, abilities or even acquired diseases of childhood and adulthood, so it is important to always recall in the context of any talk on the genome that environment plays a critical and essential component, as well.
Human Genome Perspective
Scientists have known since the mid–1950’s that we have 23 pairs of chromosomes inherited in equal numbers from our mothers and our fathers. The microscope allows these chromosomes to be divided into 2,000 bands comprising an estimated 80,000 to 100,000 genes. These 80,000 genes, each an individual bit of information for a single protein in the body, are made up of approximately three billion base pairs, or letters, of the DNA alphabet. It is the central goal of the Human Genome Project to determine the specific nature and order of each of these three billion individual bases, a number very similar to what might be found in a thousand very thick telephone books stacked top to bottom. Since a particular genetic disorder can be caused by a mutation or difference in even a single letter out of these three billion, a task set to scientists who wish to identify a particular genetic disorder is to find that single difference out of three billion, or that single letter out of the thousand telephone books that is incorrect or in error, and the outcome of which can be a serious or even fatal genetic disorder.
The genome project itself is an international effort, one that is funded by a variety of commercial, philanthropic and governmental agencies, and whose timetable for completion, at least in terms of the DNA sequence itself, has been moved up time and time again over the last several years. What had once been a final goal timeset of approximately the year 2010 is now estimated to occur perhaps even as soon as sometime later this year. These international efforts have brought together an army of scientists to develop new technologies, new ways of understanding and manipulating information and indeed new ways of working together to achieve this goal.
Funding for this comes from a variety of sources as noted, and in the current year, over 300 million dollars of the federal government’s funds support projects through the National Institutes of Health and the Department of Energy specifically targeted at the genome project. A portion of these funds, 3-5% in most years, also goes into investigating the ethical, legal and social issues of the genome project and is as important a component as the scientific mission of the project. The rate at which genome sequencing has progressed has moved from a trickle in the early 1990’s to only approximately 10 million base pairs as little as four years ago. In the last few months, the federally funded efforts exceeded one billion of the three billion letters necessary to be defined. In just the last few weeks, the company Celera, headed by Craig Venter, has reported that it now has in hand 90% of all of the sequence of the human genome. Their group has taken a strategy which has resulted in the sequence being accumulated in short bits or stretches, and although these have yet to be assembled into a long, continuous read as one would find necessary to make any sense of a chapter or an entire book, the effort, if confirmed, is impressive and holds out the promise for a substantial portion of all the sequence being truly in hand sometime in the year 2000.
Identifying a single base pair change that might underlie a human disorder once the sequence is in hand will still be equivalent to identifying a single individual on the planet as if arriving on earth from outer space. For many years, scientists have used a variety of maps, in particular genetic and physical maps, to locate the positions of genes and mutations, and it was through this effort that our laboratory first began its extensive involvement with the Human Genome Project. An example of the importance of these kinds of efforts is reflected in the talk that Michael Welsh gave in this forum just a few years ago in describing his own work on the cystic fibrosis gene. This gene was identified by using genetic mapping strategies that have afforded clinician/scientists such as Dr. Welsh—or in the case of Kevin Campbell, with the muscular dystrophy gene—the opportunity to apply their own strengths and talents to a better understanding of the biology and physiology of that gene as a way of effecting preventions and cures.
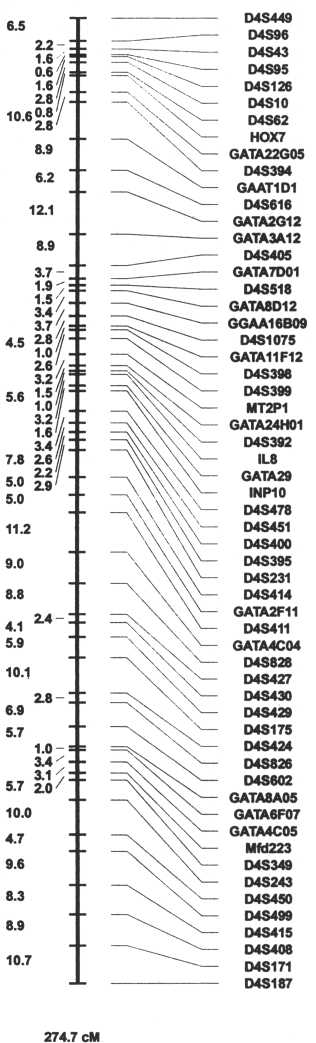
In 1993, we formed one of the human genome centers here at the University of Iowa called the Cooperative Human Linkage Center. We had six primary investigators, Geoff Duyk of Harvard, Jim Weber from the Marshfield Clinic, Ken Buetow, now at the National Cancer Institute, Val Sheffield, Robert Weir and ourselves here in Iowa. Each individual brought an area of expertise and interest into an encompassing project whose goal was to move ahead the efforts at genetic mapping and to create a series of high resolution genetic maps which, for the first time, would be truly comprehensive and easy to use. Although the project itself involved some of the most painful episodes of my scientific career, its mission was accomplished with remarkable success in that we achieved our goals well ahead of the schedule and timetable set for us by the National Institutes of Health, and in 1994, published a comprehensive set of genetic maps which remain in common and extended use today by hundreds of laboratories around the world (Murray et al. 1994). Each chromosome had its own individual map, signpost and attached reagents that made it possible for scientists in a cheap, easy and fully publicly available fashion to identify the location of their own particular gene of interest by studying families with their disorder of interest using these easily available maps and reagents.
This project was successful because of the interactive and complementary efforts of the investigators and the many graduate students, postdoctoral fellows and technicians who took part in it because of the support of the NIH. It also afforded our group the ability to now extend our interests in genetic mapping into the application of these maps to better understand birth defects, which had been from the beginning our primary interest.
Identification of the Gene for Rieger Syndrome
The application of these powerful new techniques had, in our own lab, its first specific success when in 1996 Elena Semina, a postdoctoral fellow, identified a previously unknown human gene that causes an inherited disorder comprised of glaucoma and dental abnormalities. This disorder, the Rieger syndrome, was first formally described by Herwig Rieger in 1935. He was an Austrian ophthalmologist who recognized that families in which dominant inheritance is observed had individuals who were affected with both glaucoma and dental abnormalities. While the disorder is extremely rare, we felt that it could serve as a model for other forms of glaucoma and dental anomalies, and so in the late 1980’s, we attempted to identify the causal gene.
We were assisted in this first by the work of graduate students in the laboratory who showed that chromosomal deletions in a particular area on human chromosome 4 were associated with this syndrome, and in 1992, when we were able to demonstrate conclusively that a gene for Rieger syndrome was located in the mid-portion of the long arm of human chromosome 4 (Murray et al., 1992). Subsequently, Dr. Semina and other workers in our lab set out to find the specific nature of the gene and the causal mutations in it that might lead to these disorders. We were able to make use of the technologies of the Human Genome Project in both narrowing the location in which the gene might be found, and eventually in identifying its very specific nature.
In 1996 (Semina et al.), we were fortunate to be the first laboratory on our campus to identify a previously unknown gene that was causal in a human genetic disorder, and indeed, the first gene in which glaucoma was an important phenotype. This work was only successful through the collaborative interactions of many clinicians such as Lee Alward here on our own campus, as well as other clinicians and other scientific investigators off campus who assisted us in developing the tools and reagents to find the gene.
The gene itself was of substantial interest, because it was a member of a class of very ancient genes, called “homeobox genes,” which had been shown only a decade before to play an important role in the determination of the body plan. These genes are present and conserved in almost all complex organisms and have been particularly well characterized in the fruit fly. Our gene was a member of a particular class of fruit fly genes in which abnormalities can result in body plan duplications, such as fruit flies that have two sets of wings, rather than the normal single set. Identification of this gene and its gene structure allowed Dr. Semina to characterize mutations that cause the condition and have now allowed us to extend these studies to collaborations with individuals such as Drs. Andy Russo, Roger Williamson and Baoli Yang on campus to better study the protein and to establish a mouse model for the disorder which will enable us, we hope, to eventually move on to mechanisms for treating disorders of craniofacial structures.

While I have summarized this work relatively quickly, it is important to recognize that it took us almost a decade to find this gene and that the work itself involved many people and many years of effort. We are particularly heartened by the fact that with the advances in the Human Genome Project outlined previously in this talk, a project such as this if now undertaken in the lab could likely be completed by a graduate student working alone in a year or two, enabling such students to spend the latter part of their graduate careers in understanding the functionality and clinical importance of such a disorder. In only ten years, then, we have moved from a project such as this, requiring hundreds of persons many years, as was necessary in the discovery of the cystic fibrosis or muscular dystrophy genes, to projects that could almost be completed by undergraduate students in a summer rotation. This then affords tremendous opportunities for the identification of other kinds of genes, ones that have broad implications on human health in the future.
Genes and Environment, Non-Syndromic Cleft Lip and Palate
We made use of the skills we had developed in genetic mapping to now begin to apply them to the question of what caused a relatively common birth defect called cleft lip and palate. This disorder is found in as many as one in 500 live births around the world, and because of the failure of the normal lip, palate and nasal structures to close, results in children who, as they grow, have an inability to feed properly, to speak properly and whose very disfigurement can present important social and emotional barriers to having a normal role in society. It is a complex trait in which approximately a third of the cases have other birth defects, such as heart problems or missing limbs or digits, but where 2/3 of the cases have the defect solely limited to the oral region. In these cases once the defect is repaired by the skill of surgeons such as Dr. John Canady or Dr. Janusz Bardach, the child is able to become fully functional in at least the physical sense.
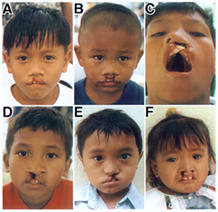
Our group has chosen to study these disorders and to try and identify the causes, both genetic and environmental. Previous work by our group and many others has demonstrated that important genetic components can include major chromosome anomalies, such as an extra chromosome number 13, many single gene or dominant or recessive conditions, and finally, that a common cause is likely to be the interactive effect of more than one gene at a time.
Environmental causes are also common and reflect the need for adequate nutrition with both vitamin A and folic acid deficiency being associated with birth defects. Exposures to environmental teratogens that can include common exposures such as cigarette smoke or alcohol, as well as more unusual exposures such as to a commonly used anticonvulsant drug, Phenytoin, can also result in a child being born with a cleft of the lip or palate.
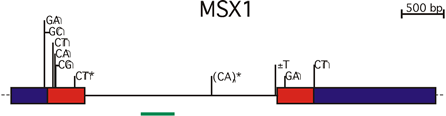
Beginning in the 1980’s, our lab has been extensively involved in trying to better understand how these defects arise, with an initial emphasis on a study of genetic factors. To carry out these studies, we identify candidate genes. These are genes which are chosen because they are expressed in the tissues of the developing face or palate or because defects of these genes in animal models such as the mouse can result in clefts of the lip or palate. Once a gene is identified, our laboratory would undertake an extensive molecular characterization, often requiring the work of one or two technicians and graduate students or postdocs for one or two years, before we would even have in hand the necessary information to carry out our genetic studies on the families. When this molecular characterization was completed, mutation detection or searching for the defects in the gene, in tens, hundreds or even thousands of individuals could be undertaken. The advances in the genome project, particularly the sequence that is already appearing in our hands and on computer screens even as we speak, has now greatly minimized the characterization step of this project so that we can move directly from selection to mutation detection, taking years off a project and allowing us to study not just one or two candidate genes but tens, or even hundreds. In an accumulation of work performed in our laboratory over the last decade and a half, beginning with Holly Ardinger and culminating with the work of Andrew Lidral in the last few years, we’ve identified several genes, two of which are shown in the figures below. One is the MSX1 homeobox gene, a gene important in developmental regulation, which shows important statistical association with cleft lip and palate in humans. This finding is validated by the fact that mutations in this gene in the mouse result in clefts in the mouse, and the gene is expressed in the critical tissues of facial expression.
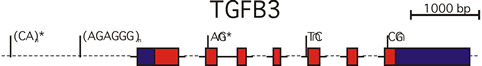
Similarly, a growth factor gene, called transforming growth factor beta 3, also has a strong statistical association with cleft lip and palate in humans, and similarly, inactivation of this gene in the mouse results in a cleft condition, and its expression is also found in that same area.
Identification of this work made use primarily of data collected from populations identified in Iowa, many of these accumulated under the direction of Dr. Paul Romitti of our Birth Defects Registry. This Iowa base afforded us the opportunity to look at genetic factors, and also to examine environmental factors; Ron Munger and other colleagues collected environmental information from the mothers and fathers of affected children. At the same time, we worked in collaboration with colleagues in Denmark, California and elsewhere around the world, and have demonstrated a strong association of cleft lip and palate with smoking, alcohol and vitamin deficiency. Many of these studies have been validated by other groups independently, and it is now well established that smoking and alcohol are strong predisposers to cleft lip and palate in humans. When combined with genetic risk factors, the risk of a parent can rise by as much as a factor of 20 for having a child with a cleft so that where a baseline risk might be 1:1000, a mother who both drinks alcohol and has a genetic risk factor could have one chance in 50 of having a child with a cleft lip or palate (Lidral et al., 1998; Romitti et al., 1999).
While these studies have been rewarding and have pointed out important risk factors, I’ve also been fortunate to become involved with a number of studies that take place outside of the United States. Some of this involvement has occurred under the direction of a volunteer medical group called Operation Smile, formed by Bill and Kathy Magee. Beginning in the late 1980’s, I’ve been fortunate enough to travel almost yearly to the Philippines to carry out these studies. Our work in the Philippines has been characterized very similarly to our work here in that the spirit of cooperation and collaboration has allowed us to progress rapidly toward important findings which we hope will set the stage for prevention and intervention trials to begin soon. The Philippines is a classic tropical island often described as a paradise, but in fact, the majority of the population lives in extremely indigent and deprived conditions. Our strategy there has been to partner with the volunteer organization Operation Smile, and local non-governmental organizations, such as the Health Organization for the Poor Enterprise (H.O.P.E.) Foundation, to work with local hospitals, authorities, public health officials and the federal and provincial government so that we can bring together groups of experts who have a good understanding of the people and their needs that allow us to move forward on these projects. We’ve been able to establish a set of field workers supervised by Sandy Daack-Hirsch. After our yearly missions to the Philippines, they continue to collect information as well as provide services, particularly to families who have newborn or young children with clefts and where the basic skills of providing adequate nutrition are often unavailable.
Our missions are characterized by an intensive 3-4 day period of screening in which hundreds of children and their families are examined by a variety of medical specialists to determine whether they are candidates for surgery. During this time, we also identify individuals with cleft lip and palate, take family histories and obtain blood samples from those families willing to participate in our studies. The presence of literally hundreds of children and young adults with unrepaired cleft lip and palate is an overwhelming sight, and one that never fails to move even those of us who have seen it many times before. The human tragedy of this disorder is brought home time and again by the stories the families and children tell us of how they have been ostracized, sick and unable to identify funds to have an operation performed which in the United States would never be denied to even the poorest of children. Many of the children we see are clearly beautiful, both inside and on the outside, and the fact that they are unfairly denied these treatments is difficult to understand, particularly when we return home to a land in which we are surrounded by so much wealth and abundance.
To initiate our studies there, we have performed epidemiologic as well as genetic and environmental studies. We have established recurrence risk data and also incidence data which has been collected in a number of hospitals in the Philippines in collaboration with their own investigators. The collection of blood samples has allowed us to carry out genetic studies similar to those that we carried out in Iowa.
| CL/P Incidence by Socioeconomic Status (SES) | ||||
|---|---|---|---|---|
| Study | Total Birth | No. CL/P | Rate/1000 | |
| Chung, 1987 | 48,729 | 71 | 1.46 | |
| Lasa, 1989 | 32,343 | 28 | 0.86 | |
| Croen, 1998 | 61,012 | 61 | 1.00 | |
| Total | 142,084 | 160 | 1.12 | High SES |
| Lasa, 1989 | 207,134 | 262 | 1.26 | |
| Murray, 1997 | 54,939 | 107 | 1.95 | |
| Total | 262,073 | 369 | 1.41 | Low SES |
Unlike our Iowa studies in which important genetic factors have been identified, we have failed to identify similar factors in the Philippines population, despite the fact that we have actually been able to study hundreds of more affected cases there, giving us much greater power for detection than was possible through the studies in Iowa where the population is much smaller and the rate of clefting much lower. These findings suggested to us that environmental components might be even more important in the Philippines than they are in Iowa, and that we could then identify these by looking for them specifically.
| Philippine Candidate Gene Studies | ||
|---|---|---|
| Gene | No. Studied Case / Control |
p value |
| TGFA | 650 / 775 | 0.37 |
| TFGB3 | 440 / 280 | 0.92 |
| MSX1 | 630 / 740 | 0.26 |
Working closely with Ron Munger, who was formerly in our College of Public Health and is now at Utah State University, we have collected dietary information, as well as data on vitamin levels. As I mentioned previously, the Philippines is a land in which, although frequently surrounded by plenty with an incredibly rich assortment of tropical fruits and vegetables, indigent populations nonetheless often subsist on an extremely monotonous diet. A typical meal for most indigent people consists of rice and dried fish, with often little supplementation from vitamin-rich vegetables or fruits, which although available may not be culturally endorsed. By collecting blood samples from such children, we have now been able to establish that vitamin B6 and folic acid deficiency seem to be associated with clefting in the Philippines, and our goal is to now extend these studies to provide us a framework for beginning vitamin interventional studies, with the hope that this can lead to not only better treatment but in fact, prevention.
At the time that we are carrying out these studies, we have frequent opportunities to have contact with our Filipino colleagues and to meet and travel with our friends. It is quite apparent early on that cleft lip and palate is not the single most important challenge facing people in the Philippines or indeed, in most indigent countries. Indeed, birth defects are well down the list of important problems in these areas. Examples of the kinds of conditions that are often found in indigent countries and which are major problems include the lack of proper nutrition, clean drinking water, immunizations and adequate shelter and food. Immunizations for common infectious diseases which are routinely available in developed countries are still lacking in indigent countries, and you can see tetanus, measles and polio. Blood products are often purchased at the site of use and are rarely screened for the wide variety of infectious agents that might be present in our own blood supplies. Almost all surgical procedures are elective and since 90% or more of the population is unable to afford them, even highly correctable lesions, such as simple and resectable tumors, and birth defects, such as clefts, cannot be treated properly.
It has been the impact of seeing the extent of the medical and physical needs of such people in the Philippines that brings me to the final part of this discussion, which is how such things resonate with us as we study human genetics in great detail. As I noted at the beginning, each of us works on what we are best at, and what I would like to spend the last few minutes on is pointing out some of the challenges raised by the genome project and our technological abilities to undertake such studies and offering suggestions for how we might think about these things in the future.
The genome project is providing a future in which we can better understand complex diseases, better diagnose them and better undertake gene discovery processes. At the same time, our ability to do this raises important social and ethical issues, both in our own country and overseas. At home, we are challenged by concerns over genetic discrimination, and whether individuals who are identified as having traits that predispose them to genetic diseases may in fact be discriminated against by insurance companies, employers or schools who will find them too high risk to take under their wing as they might do with less-at-risk citizens. These are very real concerns, and as the genome project improves in its ability to carry out such diagnoses from vanishingly small amounts of DNA, we have the possibility that dozens or even hundreds of such tests could be performed at a price low enough to be affordable by many Americans, yet provide them with information that once in hand they find they don’t really want. While it is important to always maintain the perspective that knowledge of such disorders can often be power, we have to realize that this power can be double-edged. On one side, it will allow affected individuals to undertake preventive measures through early physical screening or even adopt preventative strategies that can lower their risk for developing cancer, high blood pressure, or diabetes; on the other side, the discriminatory factor must always been considered and thought about.
Similarly, the ability to identify such individuals with high resolution has also brought about concerns about identity in general. We have all read the popular press accounts of genetic identity surrounding popular athletic and political figures, such as O.J. Simpson, Bill Clinton, and even the long-dead Thomas Jefferson, who have had their personal lives held up to intense public, as well as sometimes legal, scrutiny. While in many cases, the purpose of these is to catch and convict “bad guys,” the same technologies are also potentially subject to great abuses and invasion of personal privacy, and the balance between these is yet to be adequately identified or achieved in our society.
At the international level, many of these same concerns continue. How such information is going to be used for the world as a whole and whether genome projects will freely identify DNA sequence in a way that can benefit all of mankind and not just a few small, very wealthy private interests are examples of issues yet to be resolved. There are ongoing concerns about patenting and licensing issues related to human genetic material, and every week one can collect numerous examples from the popular literature that reflect or refer to these fears of Americans and others. As noted above, the genome project has the potential for solving many of the world’s problems, and genome projects that involve agricultural products such as wheat, corn, soybeans, as well as animal products such as the pig and the cow, all hold great promise for improving the availability of the world’s food supply. Nonetheless, as all of you are aware from reading the paper, grave concerns, particularly in areas outside the United States about the safety of genetically modified organisms, have become rampant in the last couple of years and are already leading to proscriptions on products that have wide markets already established in the United States.
A recent article in Science described rice which has been modified to contain and express the genes for vitamin A. If you live in an underdeveloped country, vitamin A deficiency is not only endemic but is a significant cause of morbidity and mortality, particularly in the young and in pregnant women. The ability to have the vitamins delivered through a point source such as rice, which is already widely accepted as a food source, would seem a miracle. This seeming miracle, however, has to be counterbalanced and brought to resolution with the concerns about the safety of doing such genetic modification and whether in the long run these modifications might, indeed, cause more harm than good.
While I’m neither an expert on genetically modified organisms, nor do I have the time to discuss them in great detail, it is clear from any reading of the popular press that this is a major concern, not only in an agricultural state such as Iowa, but for countries all around the world, both highly developed and underdeveloped. We need to better understand through education how genetic modification can work, when it can be dangerous and when it can be beneficial, and how to balance these in some way.
Finally, in our own work, concerns about human genetic modification also come to the fore, and at any time that we discuss identifying causes of cleft lip and palate, as an example, we are also really discussing what might happen if we could at some time in the future, in some way, genetically cure or prevent this. How we as a human race will resolve this need to maintain our individuality and our individual freedoms, with the desire to prevent the tragic occurrence of both physical and mental abnormalities in our children, has also yet to be resolved.
The genome project holds out tremendous promise, and our own small amount of work in looking into the development of these genetic resources and then applying them to studies of gene-environment interactions has only highlighted to myself, and I hope to all of you even more, that at the same time we push and make progress with our scientific advances, we need to constantly thinking and evaluating how they are going to have an impact on our society and on the many different societies around the world.
In closing, I’d like to thank a number of people for getting me to the point that I’m at today in being afforded the tremendous honor of being allowed to speak in front of this community and university group. Although there are many, many people to whom I owe a tremendous debt of gratitude, including the scientists I’ve tried to mention in the course of my talk, there are a few other people I would like to highlight.
First would be Jim Hanson, who first hired me here at the University of Iowa and gave me the resources and the empowerment to begin and carry out my research studies. He himself was brought here by Dr. Hans Zellweger, whose tragic suicide in the late 1980’s made me realize that even the most wonderful person, brilliant and outstanding clinician, can have troubles that may seem too insurmountable to overcome. Hugh Morris, who is here today in the audience, was the first person at the University to be nice to me when he didn’t have to be, and I’ll always be grateful to him for taking a young, naïve Assistant Professor under his wing and showing me the way to develop into not only a scientist, but I hope, a person. It was through Hugh that I met Michael Solursh, the consummate developmental biologist, father and husband, whose untimely death just over five years ago was the single greatest blow to my personal and professional career. I will never get over the loss of Michael’s friendship and his abilities, nor will our university recover from the loss of this gifted academician. We can, however, take some solace in the fact that his wonderful daughter, Libby, is carrying on the tradition of kindness and generosity that Michael showed to me. Lastly, and most importantly, I have to, of course thank my family. My first son, Ryan, who constantly reminded me of the fact that we were incredibly lucky to have a child with no birth defects and who I’m always using in my thought processes in talking with families who have not been so fortunate as us, and my wife, Ann Marie McCarthy, who have been a constant source of support throughout my work. Ryan’s two younger siblings, Chris and Katie, continue to provide me with the constant challenges and stimulation of parenthood, which far exceed those of a scientific career. Lastly, let me thank the many individuals and organizations without whom this work could not have been possible:
| Nancy Newkirk | Scott Stadler |
| Sandy Daack-Hirsch | Yuri Trembath |
| Carla Nishimura | Elena Semina |
| Sara O’Brien | Koh Yoshiura |
| Nancy Leysens | Yoko Watanabe |
| Dan Benton | Tord Hjalt |
| Buck Huppman | Rita Shiang |
| Nancy Graf | Andrew Lidral |
| Ken Buetow | Elena Semina |
| Mary Marazita | Kevin Coppage |
| Mike Dixon | Paul Romitti |
| Kaare Christensen | Lon Barrows |
| Roger Williamson | Operation Smile |
| Baoli Yang | H.O.P.E. Foundation |
| Brian Schutte | National Institutes of Health |
| Holly Ardinger | March of Dimes |
| Wade Nichols | Centers for Disease Control |
LITERATURE CITED:
- Lidral AC, Romitti PA, Basart A, Doetschman T, Leysens N, Daack-Hirsch S, Semina E, Johnson L, Machida J, Burds A, Parnell T, Rubenstein JLR, Murray JC. Association of MSX1 and TGFB3 with nonsyndromic clefting in humans. Am J Hum Genet 63:557-568, 1998.
- Murray JC, Bennett S.R, Kwitek AE., Small KW, Schinzel A, Alward WLM, Weber JL, Bell GI, Buetow KH. Linkage of Rieger syndrome to the region of the epidermal growth factor gene on chromosome 4. Nature Genetics 2:46-49, 1992.
- Murray JC, Buetow KH, Weber JL, Ludwigsen S, Sherpbier-Heddema T, Manion F, Quillen J, Sheffield VC, Sunden S, Duyk GM, Weissenbach J, Gyapay G, Dib C, Morrissette J, Lathrop GM, Vignal A, White R, Matsunami N, Gerken S, Melis R, Albertsen H, Plaetke R, Odelberg S, Ward D, Dausset J, Cohen D, Cann H. A human comprehensive linkage map with centimorgan density. Science 265:2049-2054, 1994.
- Romitti PA, Lidral AC, Munger RG, Daack-Hirsch S, Burns TL and Murray JC. Candidate genes for nonsyndromic cleft lip and palate and maternal cigarette smoking and alcohol consumption: Evaluation of genotype-environment interactions from a population-based case-control study of orofacial clefts. Teratology 59:39-50, 1999.
- Semina EV, Reiter R, Leysens NJ, Alward WLM, Small KW, Siegle-Bartelt J, Bierke-Nelson D, Bitoun P, Zabel BU, Carey JC, Murray JC. Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RGS, involved in the pathogenesis of Rieger syndrome. Nature Genet 14:392-399, 1996.
- Watson, JD and Crick, FHC. Molecular structure of nucleic acids: A structure for deoxyribose nucleic acid. Nature 171:737-738, 1953.